
Clinical trial efficacy: REACH‑2
CYRAMZA, as a single agent, is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have an alpha-fetoprotein (AFP) of ≥400 ng/mL and have been treated with sorafenib.
SELECT IMPORTANT SAFETY INFORMATION
Impaired Wound Healing
- CYRAMZA has the potential to adversely affect wound healing. CYRAMZA has not been studied in patients with serious or non-healing wounds.
- Withhold CYRAMZA for 28 days prior to elective surgery. Do not administer CYRAMZA for at least 2 weeks following a major surgical procedure and until adequate wound healing. The safety of resumption of CYRAMZA after resolution of wound healing complications has not been established.
CYRAMZA® (ramucirumab) significantly reduced the risk of death1
REACH‑2 OS: Median—Months (95% CI)1
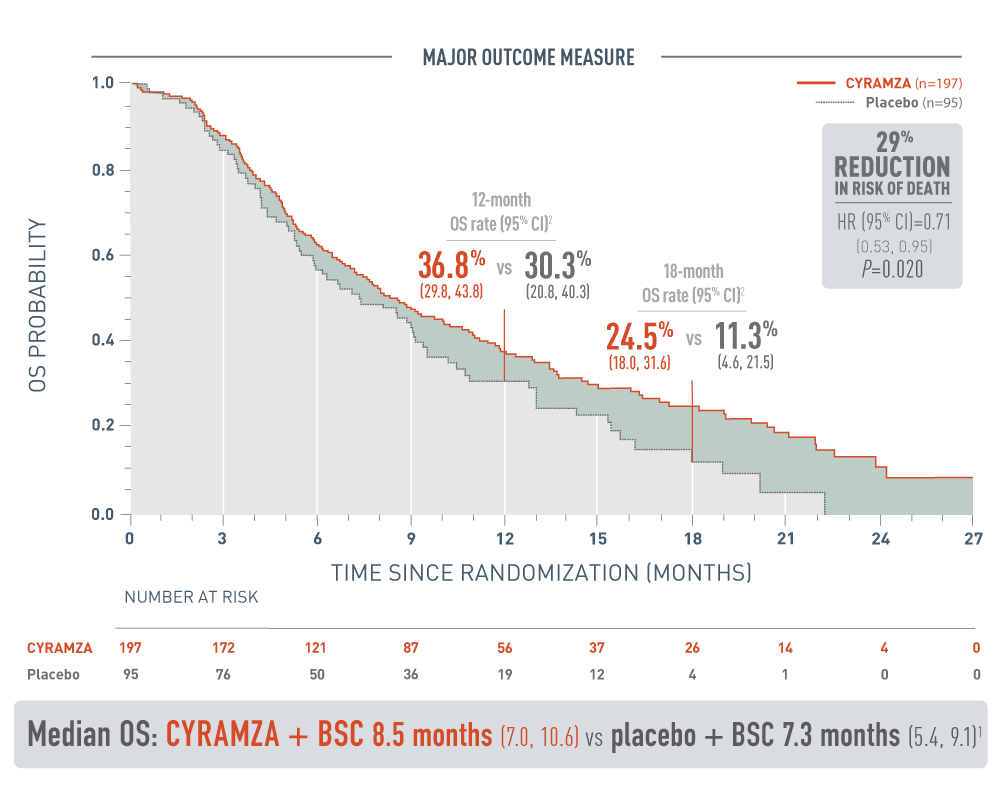
The following results for patients with hepatocellular carcinoma (HCC) who have an alpha-fetoprotein (AFP) of 400 nanograms per milliliter or higher in the REACH-2 trial are presented in a Kaplan-Meier curve.
Overall survival (OS) was the major outcome measure for the REACH-2 trial. The 12-month OS rate was 36.8 percent with a 95 percent confidence interval of 29.8 to 43.8 percent for the CYRAMZA arm (n equals 197) vs 30.3 percent with a 95 percent confidence interval of 20.8 to 40.3 percent for the placebo arm (n equals 95). The 18-month OS rate was 24.5 percent with a 95 percent confidence interval of 18.0 to 31.6 percent for the CYRAMZA arm vs 11.3 percent with a 95 percent confidence interval of 4.6 to 21.5 percent for the placebo arm. With CYRAMZA plus best supportive care (BSC), median OS was 8.5 months with a 95 percent confidence interval of 7.0 to 10.6 months. With placebo plus BSC, median OS was 7.3 months with a 95 percent confidence interval of 5.4 to 9.1 months. The hazard ratio was 0.71 with a 95 percent confidence interval of 0.53 to 0.95, and the P-value equaled 0.020. These results showed a 29 percent reduction in the risk of death for patients treated with CYRAMZA plus BSC.
- The percentage of deaths at the time of analysis was 75% (147 patients) and 78% (74 patients) in the CYRAMZA and placebo treatment arms, respectively1
REACH-2 CYRAMZA OS analysis based on prespecified patient subgroups3
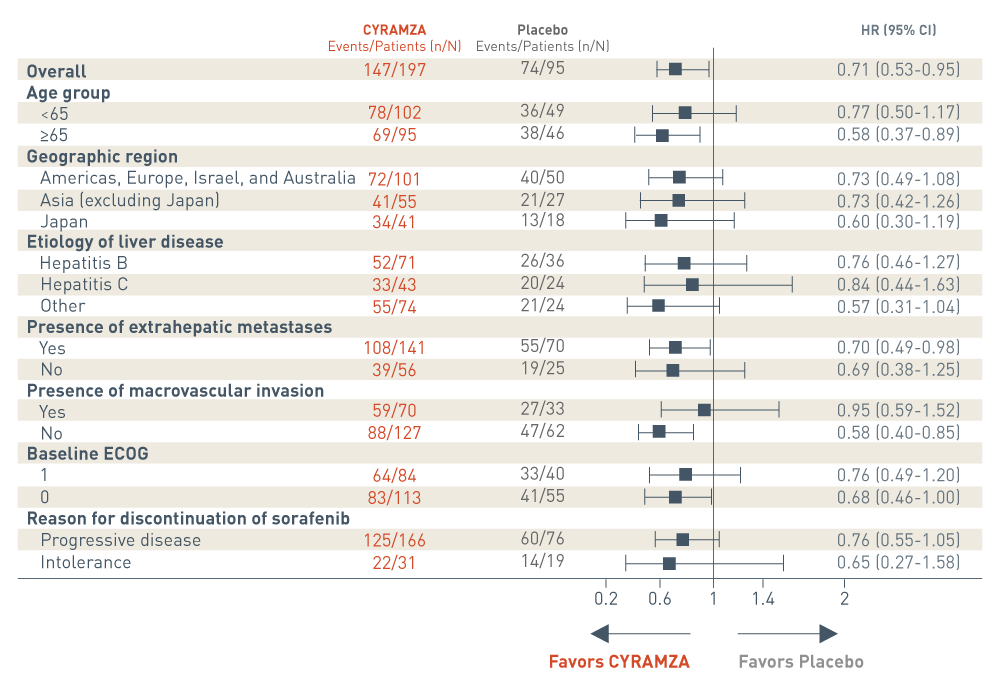
The REACH-2 trial included several prespecified patient subgroup analyses of overall survival (OS). The following results are presented in a Forest plot.
Overall, events were reported in 147 of 197 patients in the CYRAMZA arm. Events were reported in 74 of 95 patients in the placebo arm. The CYRAMZA arm was favored. The hazard ratio was 0.71 with a 95 percent confidence interval 0.53 to 0.95.
In the subgroup of patients who were less than 65 years of age, events were reported in 78 of 102 patients in the CYRAMZA arm. Events were reported in 36 of 49 patients in the placebo arm. The hazard ratio was 0.77 with a 95 percent confidence interval 0.50 to 1.17. In the subgroup of patients who were 65 years of age or older, events were reported in 69 of 95 patients in the CYRAMZA arm. Events were reported in 38 of 46 patients in the placebo arm. The CYRAMZA arm was favored. The hazard ratio was 0.58 with a 95 percent confidence interval 0.37 to 0.89.
In the subgroup of patients from the Americas, Europe, Israel, and Australia, events were reported in 72 of 101 patients in the CYRAMZA arm. Events were reported in 40 of 50 patients in the placebo arm. The hazard ratio was 0.73 with a 95 percent confidence interval of 0.49 to 1.08. In the subgroup of patients from Asia, excluding Japan, events were reported in 41 of 55 patients. In the placebo arm, events were reported in 21 of 27 patients. The hazard ratio was 0.73 with a 95 percent confidence interval of 0.42 to 1.26. In the subgroup of patients from Japan, events were reported in 34 of 41 patients. In the placebo arm, events were reported in 13 of 18 patients. The hazard ratio was 0.60 with a 95 percent confidence interval of 0.30 to 1.19.
In the subgroup of patients with hepatitis B, events were reported in 52 of 71 patients in the CYRAMZA arm. Events were reported in 26 of 36 patients in the placebo arm. The hazard ratio was 0.76 with a 95 percent confidence interval of 0.46 to 1.27. In the subgroup of patients with hepatitis C, events were reported in 33 of 43 patients in the CYRAMZA arm. Events were reported in 20 of 24 patients in the placebo arm. The hazard ratio was 0.84 with a 95 percent confidence interval of 0.44 to 1.63. In the subgroup of patients whose etiology of liver disease was classified as other, events were reported in 55 of 74 patients in the CYRAMZA arm. Events were reported in 21 of 24 patients in the placebo arm. The hazard ratio was 0.57 with a 95 percent confidence interval of 0.31 to 1.04.
In the subgroup of patients with extrahepatic metastases, events were reported in 108 of 141 patients in the CYRAMZA arm. Events were reported in 55 of 70 patients in the placebo arm. The CYRAMZA arm was favored. The hazard ratio was 0.70 with a 95 percent confidence interval of 0.49 to 0.98. In the subgroup of patients without extrahepatic metastases, events were reported in 39 of 56 patients in the CYRAMZA arm. Events were reported in 19 of 25 patients in the placebo arm. The hazard ratio was 0.69 with a 95 percent confidence interval of 0.38 to 1.25.
In the subgroup of patients with macrovascular invasion, events were reported in 59 of 70 patients in the CYRAMZA arm. Events were reported in 27 of 33 patients in the placebo arm. The hazard ratio was 0.95 with a 95 percent confidence interval of 0.59 to 1.52. In the subgroup of patients without macrovascular invasion, events were reported in 88 of 127 patients in the CYRAMZA arm. Events were reported in 47 of 62 patients in the placebo arm. The CYRAMZA arm was favored. The hazard ratio was 0.58 with a 95 percent confidence interval of 0.40 to 0.85.
In the subgroup of patients with an Eastern Cooperative Oncology Group performance status (ECOG PS) of 1, events were reported in 64 of 84 patients in the CYRAMZA arm. Events were reported in 33 of 40 patients in the placebo arm. The hazard ratio was 0.76 with a 95 percent confidence interval 0.49 to 1.20. In the subgroup of patients with an ECOG PS of 0, events were reported in 83 of 113 patients in the CYRAMZA arm. Events were reported in 41 of 55 patients in the placebo arm. The CYRAMZA arm was favored. The hazard ratio was 0.68 with a 95 percent confidence interval 0.46 to 1.00.
In the subgroup of patients who discontinued sorafenib due to progressive disease, events were reported in 125 of 166 patients in the CYRAMZA arm. Events were reported in 60 of 76 patients in the placebo arm. The hazard ratio was 0.76 with a 95 percent confidence interval 0.55 to 1.05. In the subgroup who discontinued sorafenib due to intolerance, events were reported in 22 of 31 patients in the CYRAMZA arm. Events were reported in 14 of 19 patients in the placebo arm. The hazard ratio was 0.65 with a 95 percent confidence interval 0.27 to 1.58.
- This was a prespecified exploratory analysis of the major outcome measure. The analysis was not adjusted for multiplicity or powered to detect the effect of CYRAMZA between subgroups. Therefore, no conclusions of statistical or clinical significance can be drawn.3
REACH Trial Outcomes
Median OS, the major efficacy outcome measure for the ITT population in the REACH trial, did not meet statistical significance.9
9.2 months with CYRAMZA + BSC (n=283; 95% CI: 8.1, 10.6) vs 7.6 months with placebo + BSC (n=282; 95% CI: 6.0, 9.3); HR=0.87 (95% CI: 0.72, 1.05)9
- The percentage of deaths at the time of analysis was 77.0% (218 patients) and 79.4% (224 patients) in the CYRAMZA and placebo treatment arms, respectively12
The analyses for the AFP subgroups (≥400 ng/mL and <400 ng/mL) were prespecified and exploratory without controlling for type 1 error. Observations made from these analyses cannot be considered statistically significant.
Median OS in the prespecified subgroup of patients with AFP ≥400 ng/mL showed an improvement in OS9: 7.8 months with CYRAMZA + BSC (n=119; 95% CI: 5.8, 9.3) vs 4.2 months with placebo + BSC (n=131; 95% CI: 3.7, 4.8); HR=0.67 (95% CI: 0.51, 0.90)9
- The percentage of deaths at the time of analysis was 83.2% (99 patients) and 88.5 % (116 patients) in the CYRAMZA and placebo treatment arms, respectively12
Median OS in the prespecified subgroup of patients with AFP <400 ng/mL showed no improvement in OS9: 10.1 months with CYRAMZA + BSC (n=160; 95% CI: 8.7, 12.3) vs placebo + BSC (n=150; 95% CI: 9.9, 13.1); HR=1.09 (95% CI: 0.84, 1.43)9
- The percentage of deaths at the time of analysis was 72.5% (116 patients) and 72% (108 patients) in the CYRAMZA and placebo treatment arms, respectively12
More than 500 AFP-High patients were studied in the REACH Program, adding to the body of evidence in these difficult-to-treat patients.1,5,9
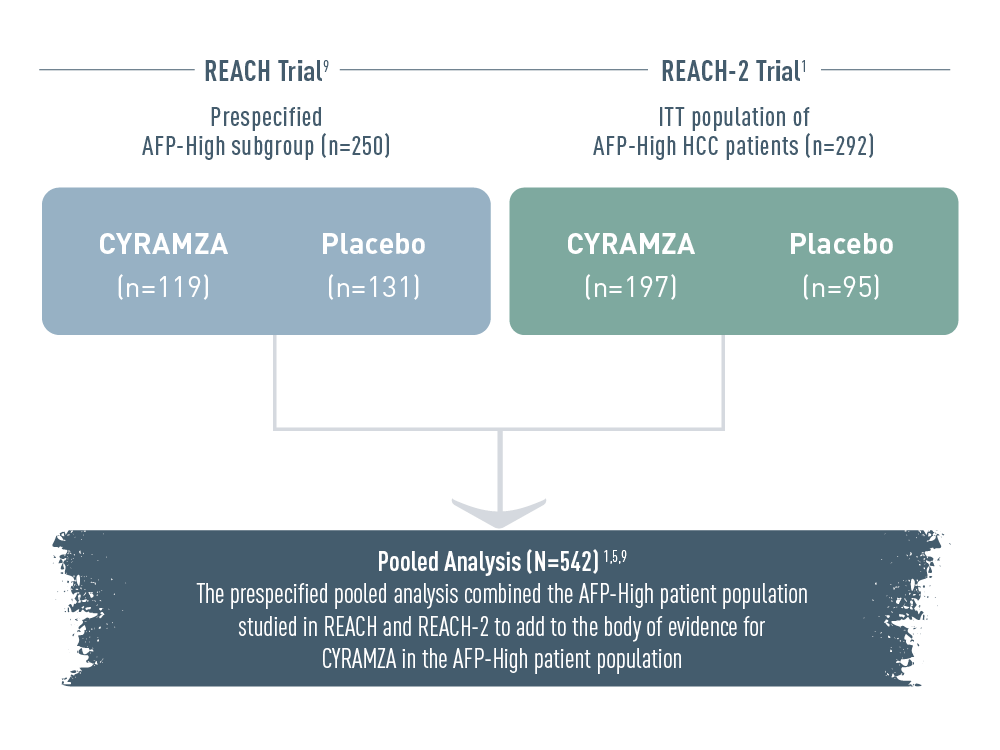
The REACH Program included alpha-fetoprotein high (AFP-High) patients from two clinical trials: the prespecified AFP-High subgroup from the REACH trial (n equals 250), and the ITT population of AFP-High hepatocellular carcinoma (HCC) patients from the REACH-2 trial (n equals 292). In the REACH trial, there were 119 patients and 131 patients in the CYRAMZA and placebo arms, respectively. In the REACH-2 trial, there were 197 patients and 95 patients in the CYRAMZA and placebo arms, respectively.
The prespecified pooled analysis combined the AFP-High patient population studied in REACH and REACH-2 (total N equals 542 patients) to add to the body of evidence for CYRAMZA in the AFP-High patient population.
The REACH Program pooled analysis was prespecified and exploratory without controlling for type 1 error. Observations made from this analysis cannot be considered statistically significant.
OS in the pooled analysis of AFP-High patients was consistent with results from REACH-2
REACH Program Pooled OS: Median-Months (95% CI)10
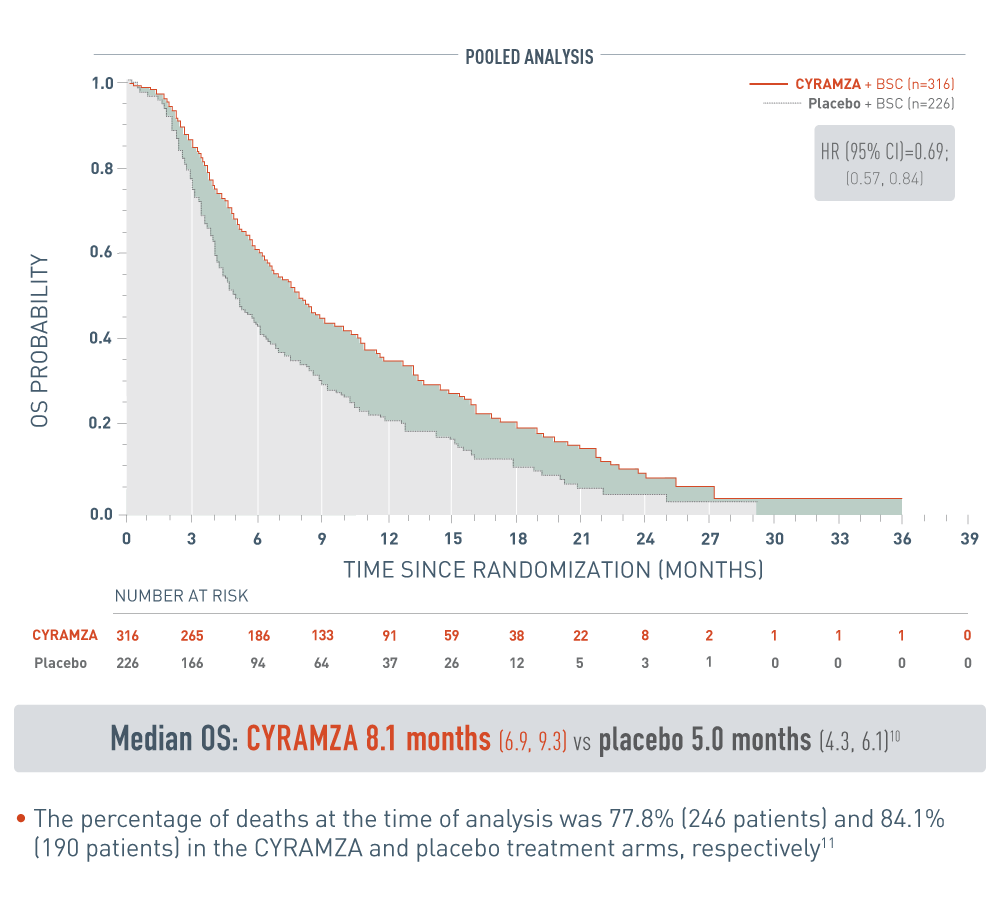
The following pooled analysis for patients with alpha-fetoprotein high (AFP-High) hepatocellular carcinoma (HCC) from the REACH program are presented with CYRAMZA plus best supportive care (BSC), (n equals 316), median overall survival (OS) was 8.1 months with a 95 percent confidence interval of 6.9 to 9.3 months, and placebo plus BSC (n equals 226), median OS was 5.0 months with a 95 percent confidence interval of 4.3 to 6.1 months. The hazard ratio was 0.69, with a 95 percent confidence interval of 0.57 to 0.84.
REACH-2 Major Outcome Measure: Median OS with CYRAMZA (n=197) was 8.5 months (95% CI: 7.0, 10.6) vs 7.3 months (95% CI: 5.4, 9.1) with placebo (n=95) (HR=0.71 [95% CI: 0.53, 0.95];P=0.0201
CYRAMZA significantly reduced the risk of disease progression or death1
REACH-2 PFS: Median Months
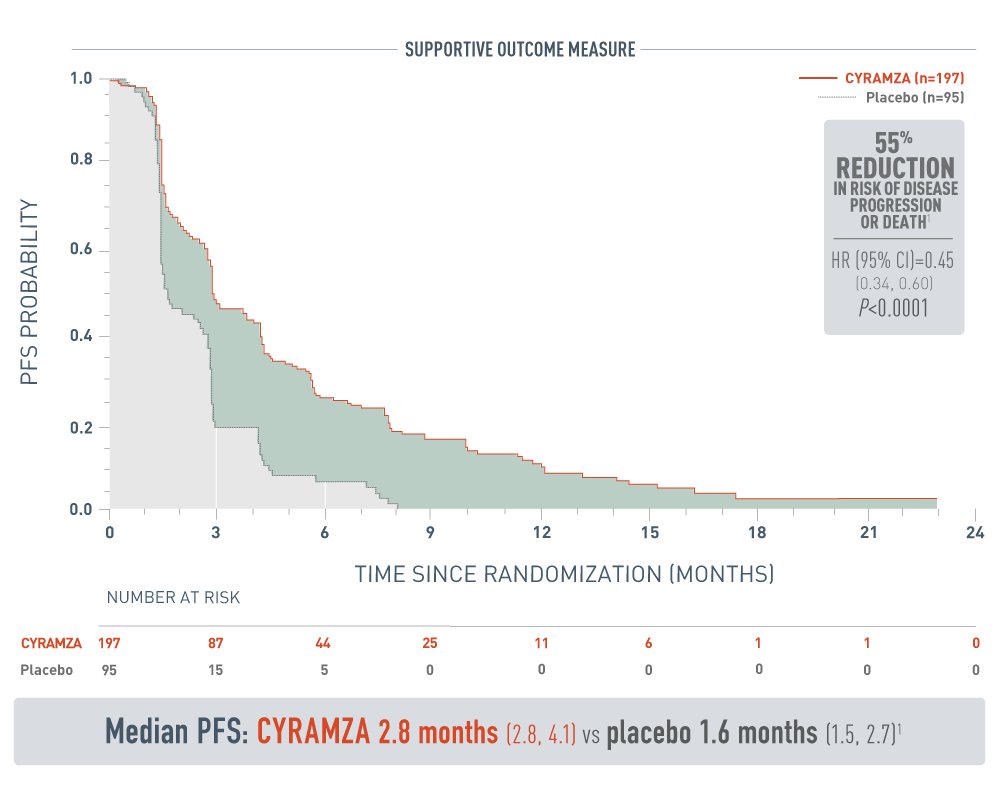
The following results from the REACH-2 trial are presented.
Progression-free survival was a supportive outcome measure of the REACH-2 trials. With CYRAMZA plus best supportive care (BSC), (n equals 197), median PFS was 2.8 months with a 95 percent confidence interval of 2.8 to 4.1 months. With placebo plus BSC (n equals 95), median PFS was 1.6 months with a 95 percent confidence interval of 1.5 to 2.7 months. The hazard ratio was 0.45 with a 95 percent confidence interval of 0.34 to 0.60 and a P value of less than 0.0001. These results showed a 55 percent reduction in the risk of progression or death for patients treated with CYRAMZA.
- The percentage of events at the time of analysis was 87% (172 patients) and 91% (86 patients) in the CYRAMZA and placebo treatment arms, respectively1
- 26 of 172 events in CYRAMZA-treated patients and 9 of 86 events in placebo-treated patients were deaths1
REACH‑2 ORR: Percentage of Patients (95% CI)
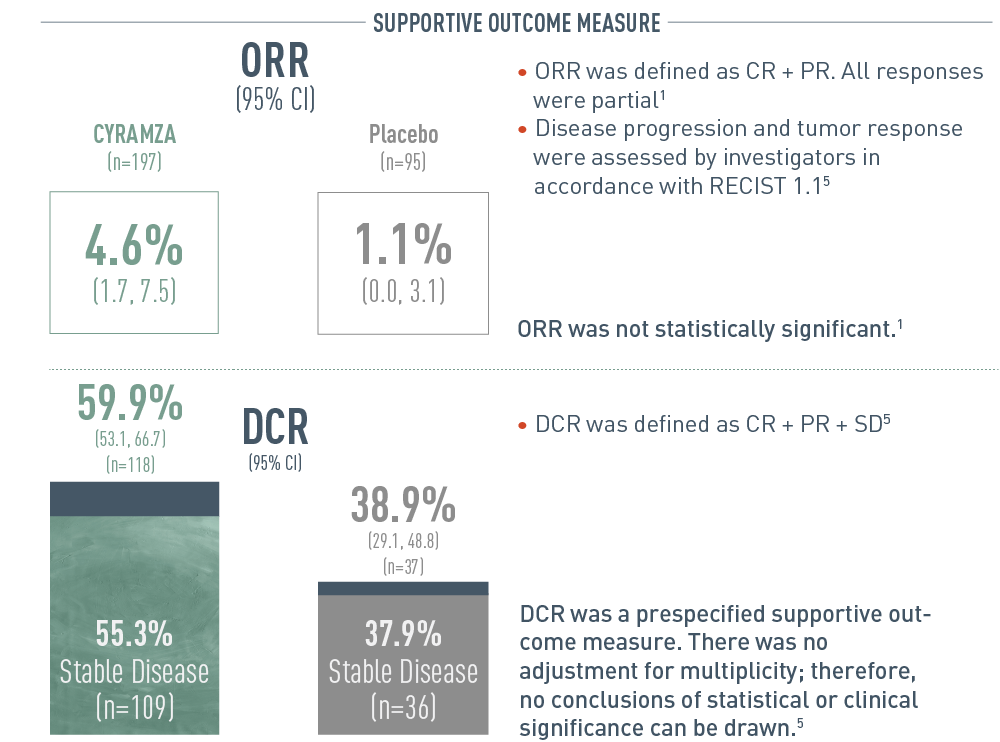
The following results from the REACH-2 trial are presented in bar graphs.
Overall response rate (ORR) was a secondary outcome measure of the REACH-2 trial. With CYRAMZA (n equals 197), ORR was 4.6 percent of patients with a 95 percent confidence interval of 1.7 to 7.5 percent. With placebo (n equals 95), ORR was 1.1 percent of patients with a 95 percent confidence interval of 0.0 to 3.1 percent. Overall response rate was defined as complete response (CR) plus partial response (PR). All responses were partial. Disease progression and tumor response were assessed by investigators in accordance Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. ORR was not statistically significant.
Disease control rate (DCR) was also a secondary outcome measure of the REACH-2 trial. With CYRAMZA, DCR was 59.9 percent of patients (n equals 118) with a 95 percent confidence interval of 53.1 to 66.7 percent. Stable disease was 55.3 percent (n equals 109). With placebo, DCR was 38.9 percent of patients (n equals 37) with a 95 percent confidence interval of 29.1 to 48.8 percent. Stable disease was 37.9 percent (n equals 36). DCR was defined as CR plus PR plus stable disease (SD). DCR was a prespecified supportive outcome measure. There was no adjustment for multiplicity; therefore, no conclusions of statistical or clinical significance can be drawn.
SELECT IMPORTANT SAFETY INFORMATION
The labeling for CYRAMZA contains warnings and precautions for hemorrhage and GI hemorrhage, including severe and sometimes fatal events; gastrointestinal (GI) perforations, a potentially fatal event; impaired wound healing; arterial thromboembolic events (ATEs), including serious and sometimes fatal events; hypertension; infusion-related reactions (IRR), including severe and sometimes fatal events; worsening of pre-existing hepatic impairment; posterior reversible encephalopathy syndrome (PRES), including fatal events; proteinuria including nephrotic syndrome; thyroid dysfunction; and embryo-fetal toxicity. CYRAMZA should be permanently discontinued in patients who experience severe bleeding, a GI perforation, an ATE, uncontrolled hypertension, severe IRR, PRES, or urine protein >3 grams/24 h or nephrotic syndrome.
The most common adverse reactions (all Grades) observed in single agent CYRAMZA-treated HCC patients at a rate of ≥15% and ≥2% higher than placebo were fatigue (36% vs 20%), peripheral edema (25% vs 14%), hypertension (25% vs 13%), abdominal pain (25% vs 16%), decreased appetite (23% vs 20%), proteinuria (20% vs 4%), nausea (19% vs 12%), ascites (18% vs 7%). The most common serious adverse reactions with CYRAMZA were ascites (3%) and pneumonia (3%).
REACH Trial Design
The phase III REACH trial evaluated the efficacy and safety of CYRAMZA vs placebo in patients with advanced HCC after prior sorafenib therapy. Major efficacy outcome measure was OS. Supportive efficacy outcome measures included PFS and ORR. The ITT population of patients was ECOG PS 0 or 1, with BCLC stage B (and no longer amenable to locoregional therapy) or C disease, and Child-Pugh A liver disease. Patients were stratified by geographic region and etiology of liver disease. Patient subgroup analysis was prespecified by baseline serum level of AFP ≥400 ng/mL. Patients were randomized 1:1 to CYRAMZA 8 mg/kg + BSC (n=283) or placebo + BSC (n=282) every 2 weeks (on days 1 and 15) of each 28-day cycle until disease progression, unacceptable toxicity, or death.9
The Major Outcome Measure for the ITT population within the REACH trial did not meet statistical significance.9
REACH-2 Trial Design
The phase III REACH‑2 trial evaluated the efficacy and safety of CYRAMZA vs placebo in patients with advanced HCC after prior sorafenib therapy and high baseline serum AFP levels ≥400 ng/mL.1 Major efficacy outcome measure was OS. Supportive efficacy outcome measures included PFS and ORR. All patients were ECOG PS 0 or 1, with BCLC stage B (and no longer amenable to locoregional therapy) or C disease, and Child-Pugh A liver disease. Patients were stratified by geographic region, macrovascular invasion, and ECOG PS. Patients were randomized 2:1 to CYRAMZA 8 mg/kg + BSC (n=197) or placebo + BSC (n=95) every 2 weeks (on days 1 and 15) of each 28-day cycle.1,5,6
For your patients with AFP ≥400 ng/mL: CYRAMZA1
The first and only biomarker-driven therapy approved for AFP-High patients with advanced HCC1,5,7
National Comprehensive Cancer Network® (NCCN®)
Ramucirumab
Specifically for patients with advanced HCC*
and AFP ≥400 ng/mL with disease progression
with a Category 1 recommendation as a subsequent treatment option in the
NCCN Clinical Practice Guidelines in Oncology [NCCN Guidelines®]8†
CATEGORY 1: Based upon high-level evidence, there is uniform NCCN consensus that the intervention is appropriate.8
NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
* Patients who have unresectable disease and are not transplant candidates; are inoperable by performance status or comorbidity, or have local disease or local disease with minimal extrahepatic disease only; have metastatic disease or extensive liver tumor burden.8
† The Category 1 recommendation is based on data reflecting use on or after sorafenib.8
CYRAMZA, as a single agent, is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have an alpha-fetoprotein (AFP) of ≥400 ng/mL and have been treated with sorafenib.

AFP=alpha-fetoprotein; BCLC=Barcelona Clinic Liver Cancer; BSC=best supportive care; CI=confidence interval; CR=complete response; DCR=disease control rate; ECOG=Eastern Cooperative Oncology Group; HCC=hepatocellular carcinoma; HR=hazard ratio; ORR=overall response rate; OS=overall survival; PFS=progression-free survival; PR=partial response; PS=performance status; RECIST=Response Evaluation Criteria in Solid Tumors; SD=stable disease.
References
- CYRAMZA (ramucirumab) package insert. Indianapolis, IN: Eli Lilly and Company; 2021.
- Data on File, Lilly USA, LLC, DOF-RB-US-0050.
- Data on File, Lilly USA, LLC, DOF-RB-US-0067.
- Data on File, Lilly USA, LLC, DOF-RB-US-0066.
- Zhu AX, Kang Y-K, Yen C-J, et al; for REACH‑2 study investigators. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH‑2): a randomised, double-blind, placebo-controlled, phase 3 trial [published online January 18, 2019]. Lancet Oncol. doi:10.1016/S1470-2045(18)30937-9.
- Zhu AX, Kang Y-K, Yen C-J, et al. REACH‑2: A randomized, double-blind, placebo-controlled phase 3 study of ramucirumab versus placebo as secondline treatment in patients with advanced hepatocellular carcinoma and elevated baseline alpha-fetoprotein following first-line sorafenib. J Clin Oncol. 2018; 36(suppl; abstr 4003).
- Llovet JM, Montal R, Sia D, et al. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599-616.
- Referenced with permission from The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Hepatobiliary Cancers V.4.2020. © National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed June 19, 2020. To view the most recent and complete version of the guidelines, go online to https://www.nccn.org .
- Zhu AX, Park JO, Ryoo B-Y, et al; REACH Trial Investigators. Ramucirumab versus placebo as second line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859-870.
- Data on File, Lilly USA, LLC, DOF-RB-US-0052.
- Zhu AX, Finn RS, Galle PR, et al. Ramucirumab as second-line treatment in patients with advanced hepatocellular carcinoma (HCC) and elevated alpha-fetoprotein (AFP) following first-line sorafenib: pooled efficacy and safety across two global randomized phase 3 studies (REACH-2 and REACH). Poster presented at: ESMO 20th World Congress on Gastrointestinal Cancer; June 20-23, 2018; Barcelona, Spain.
- Zhu AX et al. J Clin Oncol. 33, 2015 (suppl 3; abstr 232).